Activities for Society:

Epilepsy, Seizures, Treatments and Scientific Research
Epilepsia, Crisis, Tratamientos e Investigación Científica
Spanish version
Past Events
24 July 2022 | At Doca Lisboa | For General Public
25 July 2022 | At iMM | For Researchers
Raising awareness on epilepsy & Building the future of epilepsy research
with Sail for Epilepsy & Liga Portuguesa contra a Epilepsia

Sail for Epilepsy on Portuguese National news - see here
. . .
25 March 2022 | Lisbon, Portugal
Round table
"Epilepsies in a school context"
What is epilepsy? How to detect and act in case of seizures? How to communicate?
A scientist, a neuro pediatrician, a nurse and a psychologist get ready to talk!


In this round table we discussed strategies to understand, detect and deal with cases of seizures and epilepsies in a school context.
Organized as an hybrid event gathered around 20 people in person and 100 people through zoom.
Stay tuned for future initiatives like this one!

Did you know that there are over 300.000 people living with rare diseases?
Over 6000 rare diseases have been identified. 72% have a genetic whilst others are caused by infections, allergies, or environmental factors. Furthermore, 70% of genetic rare diseases start in childhood.
You can find this and more information in https://www.rarediseaseday.org/.
February is the month of rare diseases and, in 2021, the Rare Disease day is on the 28th of February.
Some of these rare diseases lead to the development of epilepsy.
During February we will dedicate some posts on raising awareness about some of these diseases.
You can check them by clicking in "Rare Diseases" in the left menu of the website!
Do you want to hear more about the research we do?
Do you want to be updated on upcoming events?

Follow us on Twitter: @epiepinet
Visit us on Facebook: EpiEpi Net
Network with us on LinkedIn: epiepinet
February 2021 - Rare Disease Month

There are over 300.000 people leaving with rare diseases and over 6000 rare diseases have been identified.
Some of theses diseases involve the development of epilepsy and, in some cases, the development of refractory forms of epilepsy which are resistante to treatment with antiepileptic drugs.
During the rest of the month we will share with you information on some of these diseases and the developments that have been made to understand the cellular & molecular mechanisms behind them.
Rett Syndrome
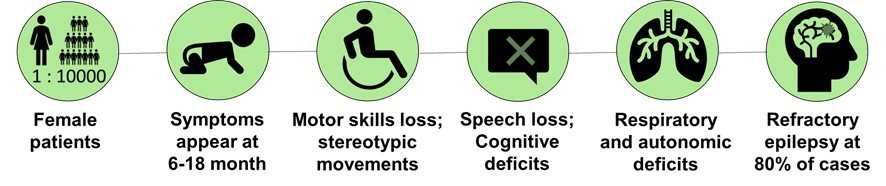
Rett Syndrome is a rare neurological disorder that affects normal development and function of the brain.
It affects mostly, but not exclusively, women. With an incidence of 1 case in each 10 000 live births, it is one of the major genetic causes of intellectual disability in girls.
Rett Syndrome is characterized by an apparent normal early development until 6-18 months of life, followed by a regression with loss of motor, speech, and cognitive skills, as well as deficits in respiratory and autonomic development.
Furthermore, around 80% of Rett syndrome patients develop refractory epilepsy.
EPILEPSY:
Epilepsy is a neurological disease characterised by the occurrence of seizures. These seizures are caused by an abnormal firing of brain cells, the neurons. In most cases, the occurrence of seizures can be reduced or completely stopped by following treatment with antiepileptic drugs. In other cases, seizures seem to be resistant to those drugs. Those are known as refractory epilepsies.
In more than 90% of the cases, Rett Syndrome is caused by mutations in the MECP2 gene.
Located in the X-chromosome, the MECP2 gene encodes for a protein that regulates the expression of other proteins during different phases of brain development.
One of the Strategic Research lines at EpiEpiNet is to study how the development and function of neurons is impaired in Rett Syndrome.
In 2020, EpiEpiNet researchers published 3 research papers on Rett Syndrome. Do you want to read about this research? Check the rest of the post...
![]()
1. Research arcticle published in Frontiers in Cell & Developmental Biology:
- Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids -
One of these studies resulted from collaboration between researchers from iMM and Instituto Superior Técnico, in Portugal, and the Universities of California and Rotterdam.
In this work researchers focused on developmental features of the disease.
For that they used brain organoids as a model.
BRAIN ORGANOIDS:
Sometimes referred to as “mini brains” in the non-academic world, brain organoids are a recently developed tool to study mechanisms of human brain development in the laboratory. Even though it still requires some technical improvements, the use of brain organoids can become a potent tool for personalised medicine and drug testing. One of the great advantages of brain organoids is the use of cells directly isolated from patients with specific mutations.
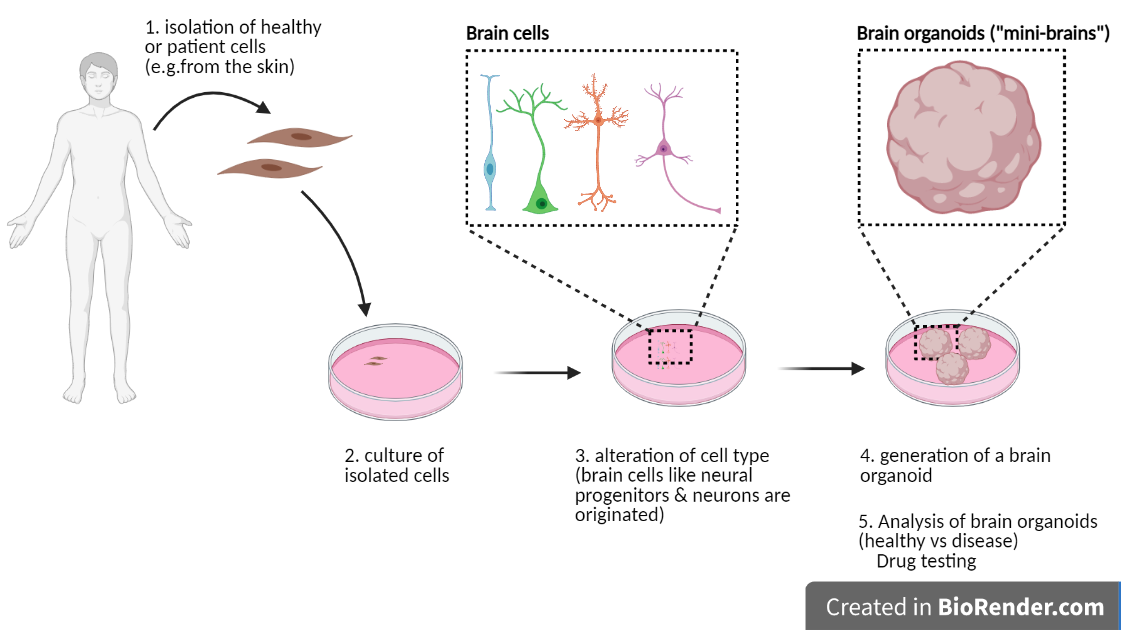
In this research work, brain organoids were obtained from cells of Rett Syndrome patients carrying a mutation in the MECP2 gene.
To interpret this study, it is important to understand some basic features about the brain development:
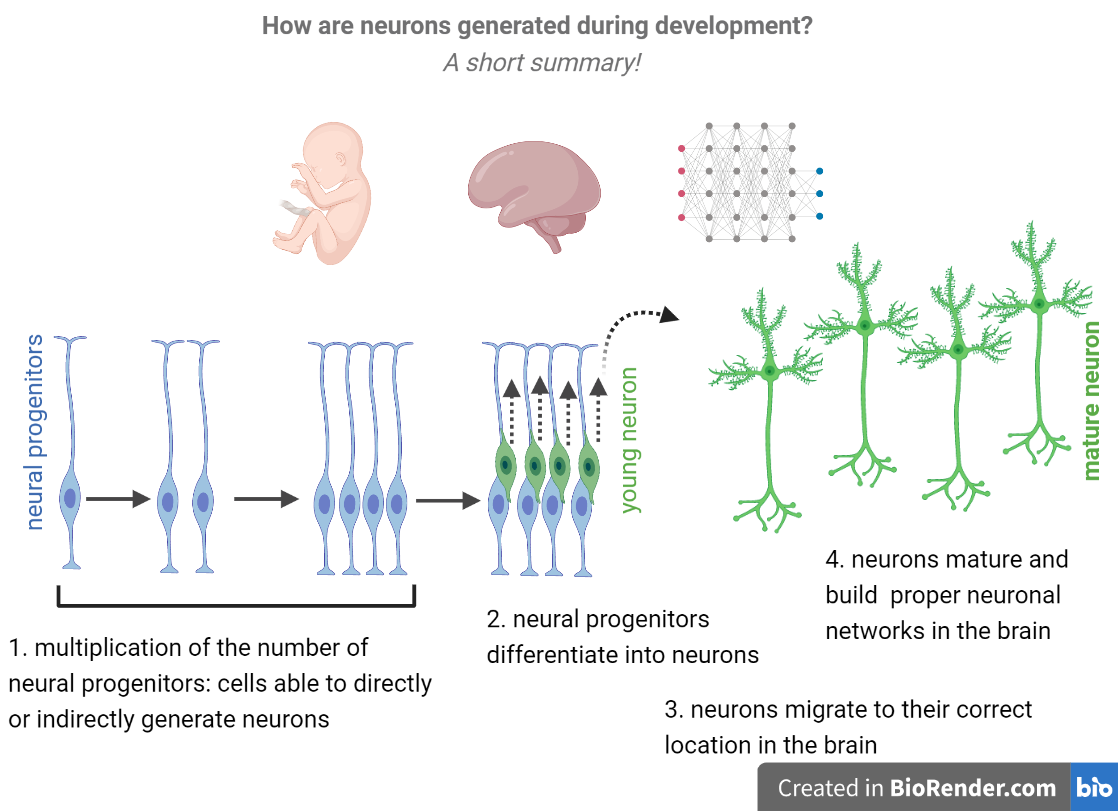
1. During embryonic brain development a group of cells known as neural progenitors divide rapidly and, later on, differentiate into neurons.
2. After being formed, neurons migrate, mature and form networks in the brain.
3. Once differentiated, neurons can no longer divide and, therefore, do not originate more neurons. This means that, if neural progenitors differentiate too early into neurons, there is a premature loss of neural progenitors from the brain and, in the end, less neurons.
What researchers found is that, in brain organoids derived from Rett Syndrome patients, certain neural progenitors differentiate too soon into neurons.
Furthermore, while some types of neurons face problems during maturation, others have problems in moving towards their final correct destinations.
These defects may contribute to some of the neurodevelopmental defects observed in certain Rett Syndrome patients.
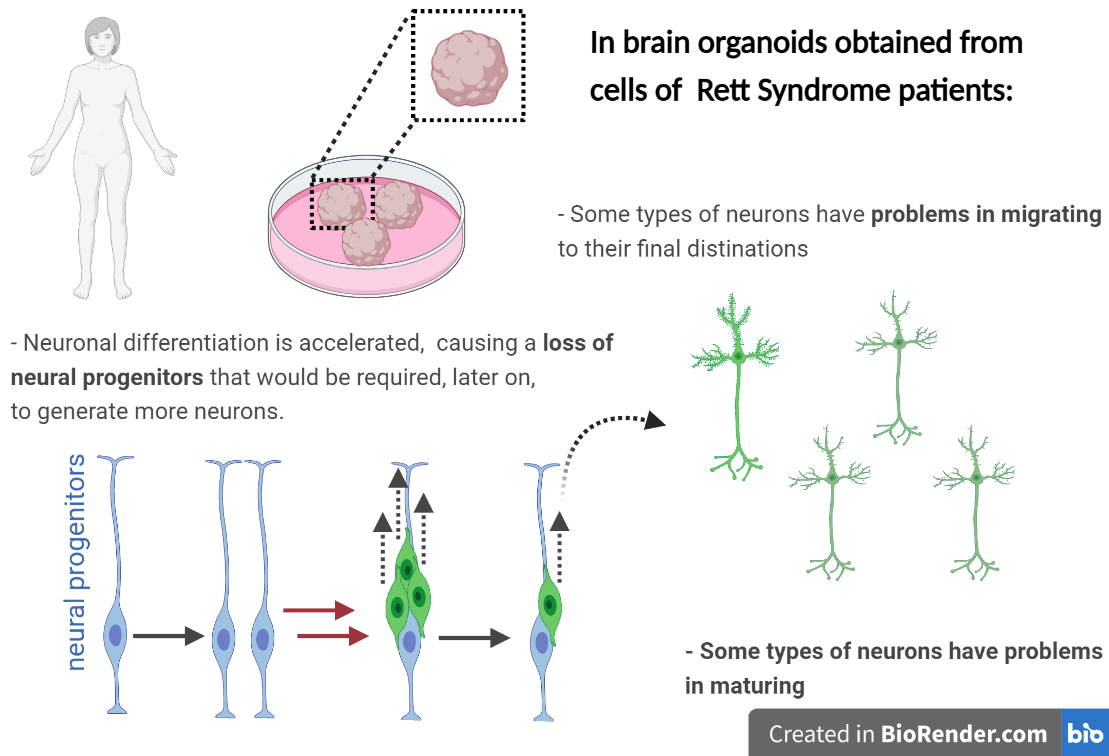
Apart from the mechanistic breakthroughs, this study brings technical advances in the development of brain organoids to study Rett Syndrome. These advances are essential to allow the use of brain organoids for disease characterization, personalized medicine and drug testing.
![]()
2. Reasearch arcticle published in Neurobiology of Disease:
- Impairment of adenosinergic system in Rett syndrome: Novel therapeutic target to boost BDNF signalling -
Another of these studies involved researchers from Hospital San Joan de Deu, in Spain and Instituto de Ciencias Biomédicas Abel Salazar and iMM, in Portugal.
This work used animal models of Rett Syndrome to focus on how neurons and neuronal activity are affected in this disease.
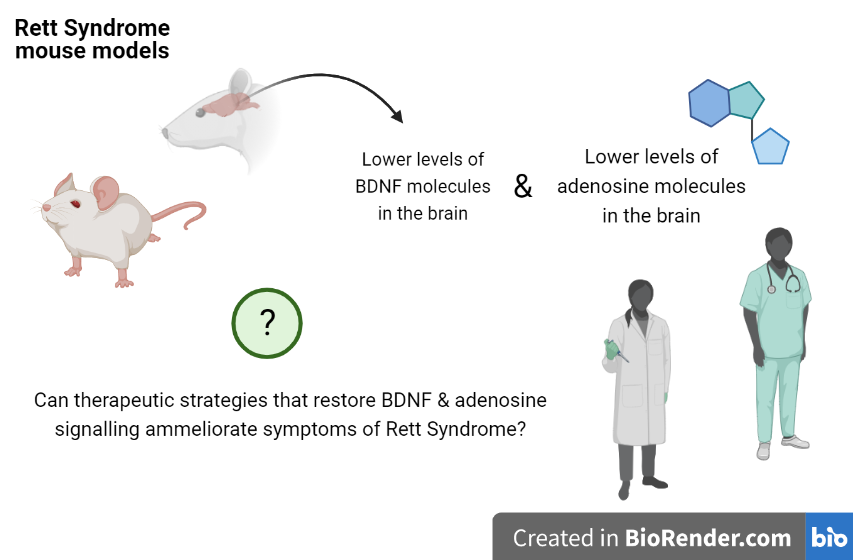
Researchers studied how two important molecules involved in neuron function have an impact in Rett Syndrome. These molecules are BDNF (brain derived nerve factor) and adenosine.
BDNF is crucial for the development of neurons and their ability to stablish communication points with other neurons in the brain. In line with previous studies, authors show that the amount of BDNF and other related proteins are reduced in the brain of mouse models of Rett Syndrome.
This confirmation is particularly important: other groups had shown that manipulation of BDNF levels can ameliorate symptoms in mice with Rett Syndrome. Direct translation to human treatment is still hard. However, molecules that could potentiate BDNF levels or action would be a great step towards that direction.
Following that possibility, researchers focused on adenosine.
ADENOSINE:
Adenosine exists naturally in our brain and is involved in regulating the activity of neurons and in many biological processes like sleep and awareness. Indeed, it acts as a natural anti-epileptic molecule in the brain ensuring correct levels of brain activity. Studies at iMM were pioneers to show that several actions of BDNF in the brain are dependent on adenosine.
What iMM researchers now found is that mouse models of Rett syndrome also have lower levels of adenosine.
This is an important finding considering that some research groups are testing the use adenosine as a novel therapeutic strategy for epileptic seizures.
Therefore, some potential implications of this work could be the possibility of using adenosine-based strategies for the treatment of Rett Syndrome patients.

3. Reseach arcticle published in Neuroscience:
The brain is composed of around 100 million neurons, able to conduct information in the form of electrical signals. These neurons are organised in a complex network where they make connections with other neurons. These connections take place at specific places known as synapses. It is thought that in the adult brain there more than 125 trillion of these connection points.
- At synapses, when a neuron passes information to another neuron, it converts the electrical signal to chemical signal through the release of little molecules known as neurotransmitters.
The recognition of these chemical signals by other neurons occurs in most cases in the border of the cell (at the cell membrane) and is mediated by proteins known as receptors.
At the synapses, neurons also have other proteins known as ion channels. Ion channels control the movement of electric charges from the inside to the outside of the cell, and vice versa. When a neurotransmitter binds to a receptor, it can cause the opening or closure of ion channels creating (or blocking) new electric signals in the second neuron.
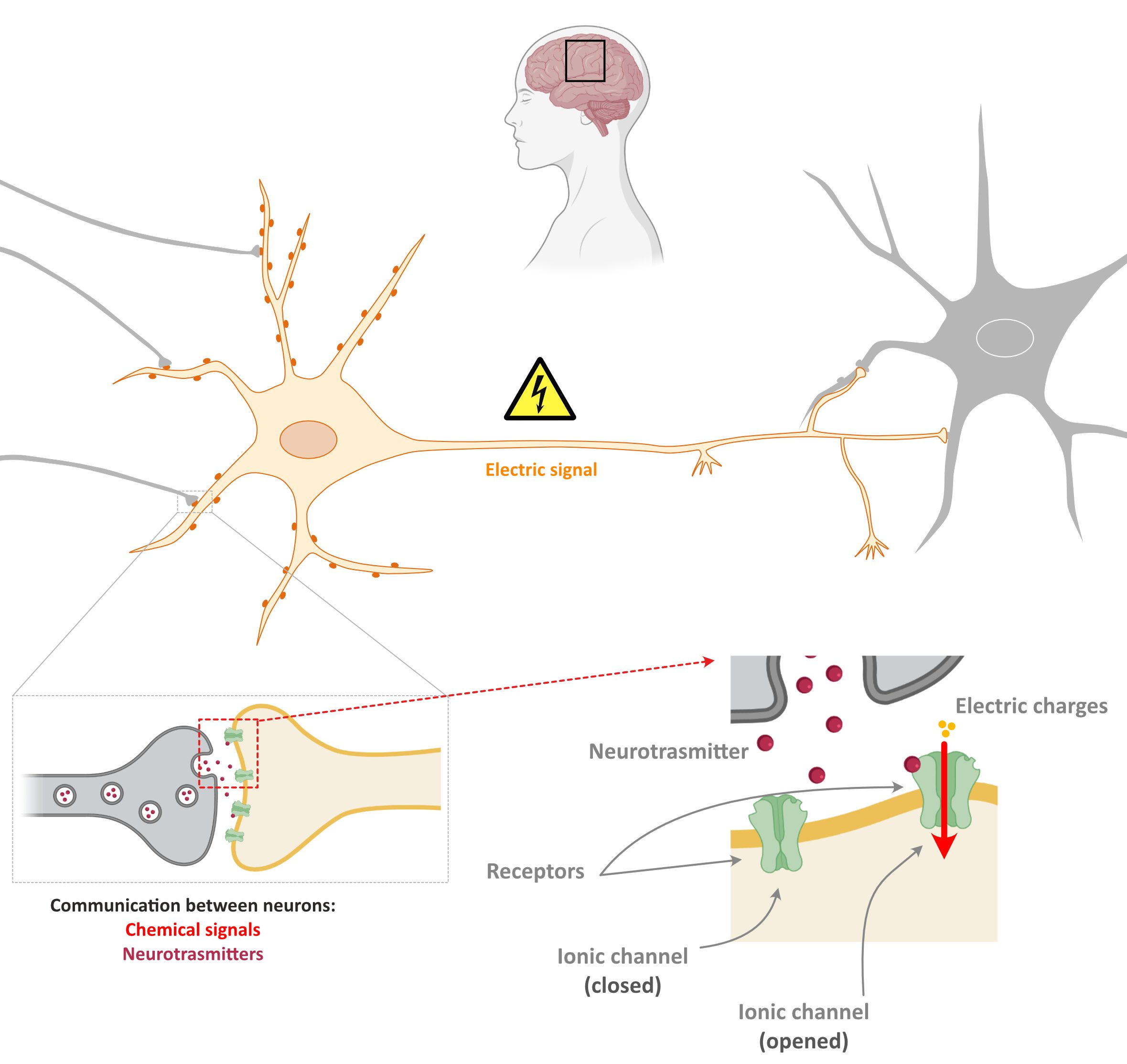
This is the principle on how information in the brain is transmitted!
In several diseases like the Rett Syndrome, there is an unbalance between the different signals, namely between inhibitory and excitatory signals which can lead to the occurence of seizures.
In a collaboration study from EpiEpiNet partners at iMM, Amsterdam Medical Centre and Sapienza University of Rome, an innovative technique, developed some years ago by Ricardo Meledi, was used to study the dysfunction of a key inhibitory receptor and channel in Rett Syndrome.
...
At EpiEpiNet we believe that, going step by step, the scientific community will achieve a better understanding of the biological causes and symptoms of Rett Syndrome. This knowledge will, hopefully, pave the way for development of so needed therapeutics for this disease. EpiEpiNet is proud of taking part in this process.
February 2021 - Rare Disease Month

There are over 300.000 people leaving with rare diseases and over 6000 rare diseases have been identified.
Some of theses diseases involve the development of epilepsy and, in some cases, the development of refractory forms of epilepsy which are resistante to treatment with antiepileptic drugs.
During the rest of the month we will share with you information on some of these diseases and the developments that have been made to understand the cellular & molecular mechanisms behind them.
MAND
MAND is a term that describes a group of very rare neurodevelopmental disorders involving alterations in a specific genomic region in the chromosome 2: the 2q23.1 locus.
MAND is a dominant disorder which means that genetic alterations in any of the two copies of the chromosome 2 might cause disease.
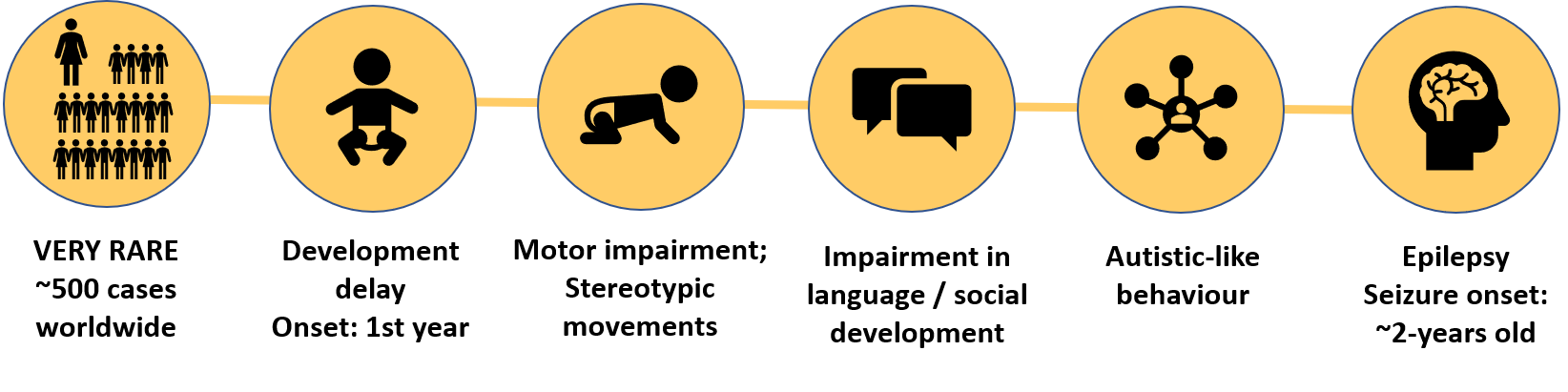
In more than 80% of the cases, MAND is associated with:
- Developmental delays starting within the first year of life
These include motor delays, an impairment in language development and social development delay. - Occurrence of Stereotypic repetitive behaviours
- Sleep disturbances
- Microcephaly
- Autistic-like behaviour
- Seizures: The onset of seizures usually occurs around 2 years of age. In some of the cases refractary epilepsy is observed. This is, epilepsy that is resistant to treatment with anti-epileptic drugs. Seizures have not been well characterized in this population, however, most individual exhibit only one seizure type.
Some of MAND symptoms are similar to those observed for other Syndromes like Rett (e.g. the stereotypic hand behaviour seen in Rett Syndrome patients), Angelman, X-fragile, and others.
Unfortunately, sometimes these shared features, together with the fact that MAND is very rare, can lead to misdiagnosis of the disease.
To manage and treat MAND patients it is very important to take an individual-oriented multidisciplinary approach which includes: medical genetics, speech & language therapy, occupational & physical therapy, child development, neurology, nutrition and ophthalmology.
Information on MAND and the biological causes behind it is scarce. Still, the academic community has made some advances towards it understanding.
Here you can find a good scientific review on MAND done by researchers from California and Texas, in the USA:
https://www.nature.com/articles/ejhg201635
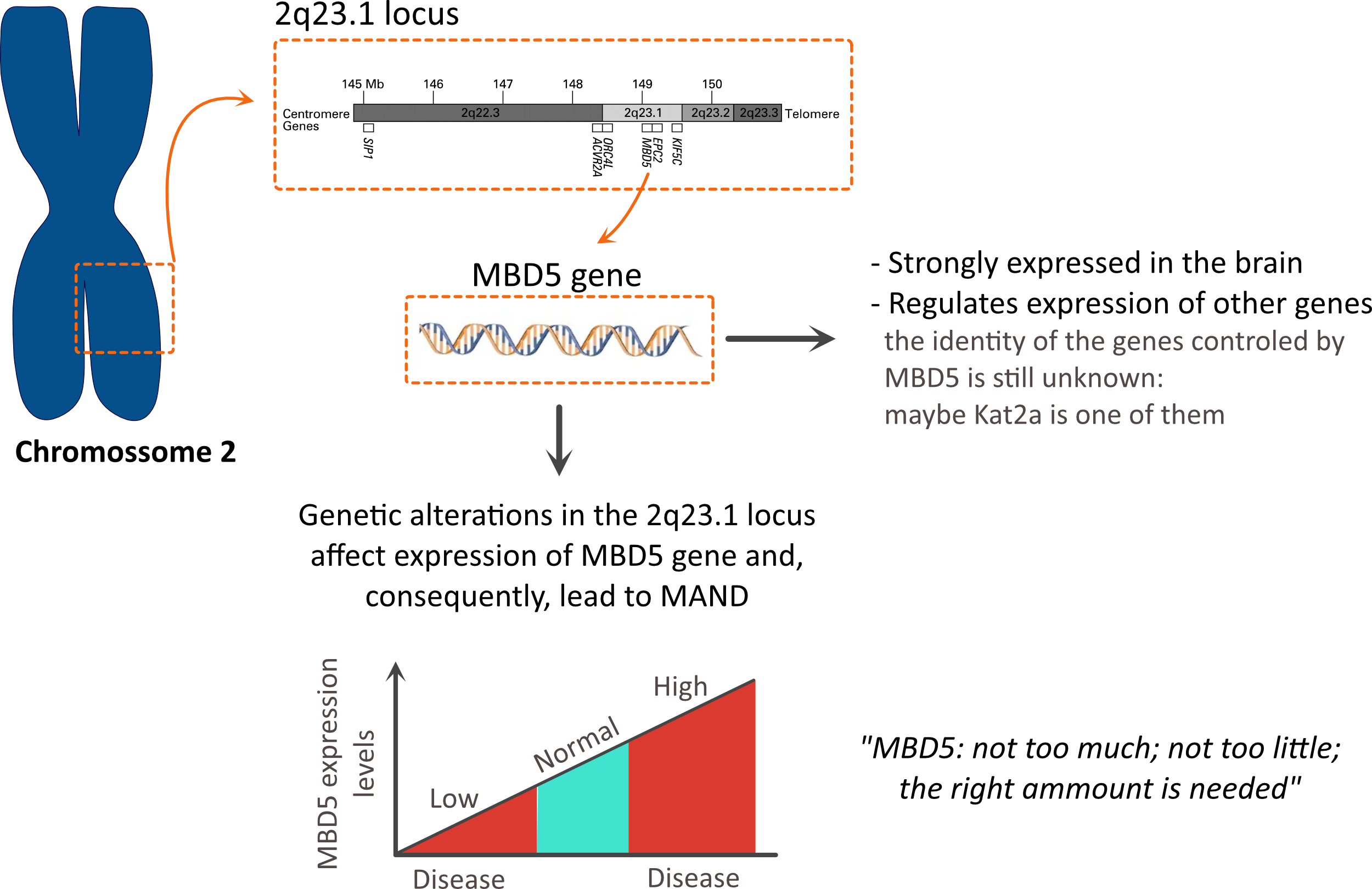
The term MAND stands for MBD5-associated
neurodevelopmental disorder
The origin of the name pops up from the fact that genetic alterations in the 2q23.1 region causes changes in MBD5: in some cases, an increase in the levels of MBD5 is observed; in other cases, a reduction in the levels of MBD5 is observed; both increase, or reduction is linked to disease.
This is a clear example in biology where tight control of the expression of genes is essential during development (not too much; not too little but the right amount).
But why is MBD5 so important?
- MBD5 is a protein that acts as a transcription regulator.
This means that it controls the expression of other proteins: something similar to what MECP2 does (the gene involved in Rett Syndrome). Indeed, MBD5 and MECP2 belong to the same family of proteins.
However, which genes are controlled by MBD5 is still unknown.
- Furthermore, high levels of MBD5 are found in the brain.
To understand the biological mechanisms behind diseases and develop appropriate therapies it is important to count on animal models to study these diseases.This is also the case for MAND.
Indeed, in the last years several mouse models have been generated.
These models use genetic engineering to alter the levels of MBD5 in the mouse organisms.
In some of the cases, these mouse models recapitulate well the features observed during human development in MAND patients.
Even though a better characterization of these models is needed, the existence of these models is already good news: they a very potent tool to get a better understanding on MAND.
Another question that needs to be clarified by researchers is if MBD5 is the only protein that is impaired in MAND.
For example, in the proximity of the 2q23.1 locus there are also other proteins like ORC4, KIF5C and EPC2 which play an important role during development and brain function.
Whether these proteins could contribute to the features observed in MAND is still unknown.
…
At EpiEpiNet we believe that, going step by step, the scientific community will achieve a better understanding of the biological causes and symptoms of rare diseases involving refractary forms of epilepsy. This knowledge will, hopefully, pave the way for development of so needed therapeutics for these disease. EpiEpiNet is proud of taking part in this process.