Atividades para a Sociedade:

Epilepsia, Crises, Tratamentos e Investigação Científica
Últimos eventos:
24 Julho 2022 | Doca Lisboa | Para o Público Geral
25 Julho 2022 | iMM | Para investigadores
Informar sobre a Epilepsia
com "Velejar pela Epilepsia" - SAIL FOR EPILEPSY - e a Liga Portuguesa Contra a Epilepsia

Mais sobre a Sail for Epilepsy numa reportagem da RTP aqui
25 Março 2022 | Mesa Redonda
Auditório Cid dos Santos | Faculdade de Medicina de Lisboa & Zoom
"Epilepsias no meio escolar"
O que é a epilepsia? Como detectar e atuar em caso de crises? Como comunicar?

Uma cientista, uma neuropediatra, uma enfermeira e uma psicóloga sentam-se a uma mesa a falar. Qual será o resultado? Junte-se a nós!

Dia Internacional das Doenças raras
Existem mais de 300 000 pessoas em todo o mundo com doenças raras.
Por outro lado, foram já identificadas mais de 6000 doenças raras, cada uma com as suas características sintomáticas.
Algumas destas doenças raras estão associadas à ocorrência de crises
epiléticas e, em alguns casos, ao aparecimento de epilepsias refratárias,
resistentes ao tratamento com fármacos antiepiléticos.
Na nossa página partilhamos convosco informação sobre algumas destas doenças. Daremos uma atenção especial a novas descobertas que ajudam a compreender melhor os mecanismos celulares e moleculares por detrás de algumas destas doenças.
Quer manter-se informado sobre a investigação levada a cabo pelo EpiEpiNet?
Quer manter-se informado sobre eventos futuros?

Siga-nos no Twitter: @epiepinet
Visite-nos no Facebook: EpiEpi Net
Conecte connosco no LinkedIn: epiepinet
Fevereiro 2021 - Mês das Doenças Raras

Existem mais de 300 000 pessoas em todo o mundo com doenças raras.
Por outro lado, foram já identificadas mais de 6000 doenças raras, cada uma com as suas características sintomáticas.
Algumas destas doenças raras estão associadas à ocorrência de crises epiléticas e, em alguns casos, ao aparecimento de epilepsias refratárias, resistentes ao tratamento com fármacos antiepiléticos.
Na nossa página partilhamos convosco informação sobre algumas destas doenças. Daremos uma atenção especial a novas descobertas que ajudam a compreender melhor os mecanismos celulares e moleculares por detrás de algumas destas doenças.
Síndrome de Rett

O Síndrome de Rett é uma doença neurológica que afeta o desenvolvimento normal do cérebro.
Afeta maioritariamente, mas não exclusivamente, mulheres.
Com uma incidência de 1 caso em casa 10 000 nascimentos é uma das principais causas genéticas de défice intelectual em meninas.
O Síndrome de Rett é caracterizado por um desenvolvimento aparentemente normal até ao 6-18 meses de vida, seguido por uma regressão com perda de capacidades motoras, de fala e cognitivas, bem como défices respiratórios e autonómicos.
Cerca de 80% dos doentes com Síndrome de Rett desenvolvem ainda formas refratárias de epilepsia.
EPILEPSIA:
A epilepsia é uma doença neurológica caracterizada por uma predisposição para a ocorrência de crises epiléticas.
Estas crises epiléticas são causadas por uma atividade anormal de umas das principais células no nosso cérebro, os neurónios. Hoje, a grande maioria dos doentes consegue reduzir ou eliminar totalmente a ocorrência de crises epiléticas através do tratamento com medicamentos antiepiléticos. Num menor número de casos, as crises mantêm-se apesar da medicação e doses corretas. São as chamadas epilepsias refratárias.
Em mais de 90% dos casos o Síndrome de Rett é causado por mutações no gene MECP2.
Localizado no cromossoma-X, o gene MECP2 codifica para uma proteína que regula a expressão de outras proteínas cujo papel é importante durante diferentes fases do desenvolvimento do cérebro.
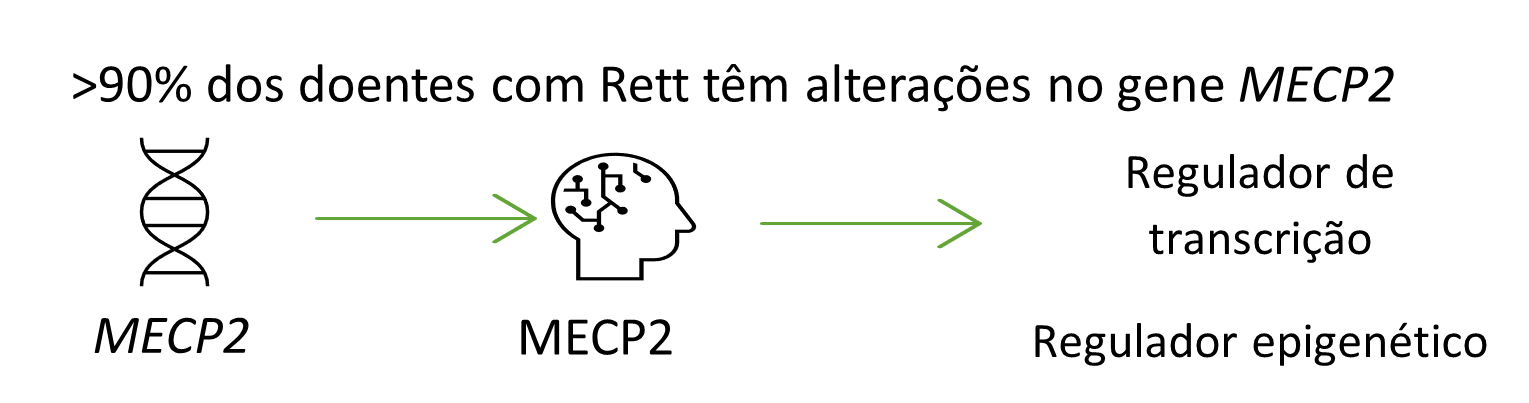
Uma das Linhas Estratégicas de Investigação da rede EpiEpiNet é o estudo do Síndrome de Rett e de que forma o desenvolvimento e funcionalidade do cérebro são afetados nesta doença.
Em 2020, investigadores da EpiEpiNet publicaram 3 artigos de revisão relacionados com o Síndrome de Rett.
Quer saber mais sobre estas investigações? Veja o resto da publicação…![]()
1. Artigo de investigação publicado na revista Frontiers in Cell & Developmental Biology:
-Utilização de organoides de cérebro derivados de células de doentes como modelo para o estudo do Síndrome de Rett-
(Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids)
Um destes estudos resulta de uma colaboração entre investigadores do iMM e do Instituto Superior Técnico, em Portugal, e das Universidades da Califórnia e de Roterdão.
Neste trabalho, os investigadores focaram-se no estudo de algumas das características da doença que se observam durante o desenvolvimento.
Para tal, utilizaram organoides de cérebro como modelo.
ÓRGANOIDES DE CÉREBRO :
Às vezes referidos como “mini-cérebros” no mundo não académico, os organoides de cérebro são uma metodologia pioneira e relativamente recente que permite o estudo no laboratório de mecanismos associados ao desenvolvimento do cérebro humano.
Apesar de necessitar ainda de alguma otimização e melhorias técnicas, a utilização de organoides de cérebro humano tem um elevado potencial como ferramenta para estudos de medicina personalizada e de testagem pré-clínica de medicamentos. Uma das grandes vantagens dos organoides é a possibilidade de utilizar células isoladas diretamente a partir de doentes com alterações genéticas que lhe são específicas.
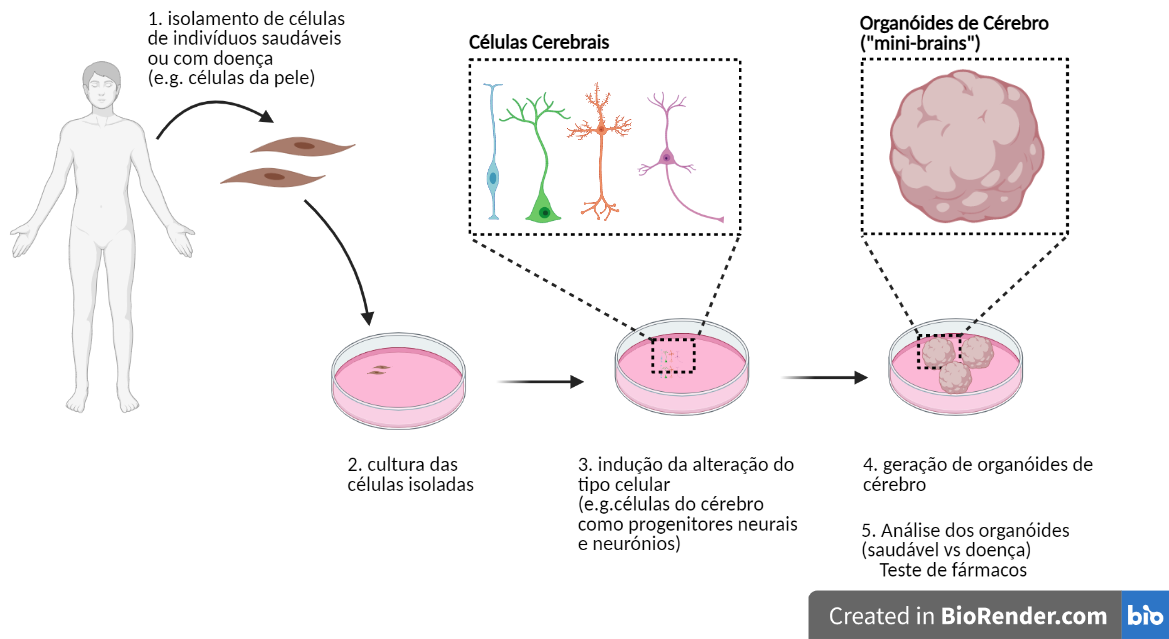
Neste trabalho de investigação foram obtidos organoides a partir de células de doentes com mutações no gene MECP2 e que sofrem de Síndrome de Rett.
Para interpretar estes resultados é importante compreender alguns dos conceitos envolvidos no desenvolvimento do cérebro humano:
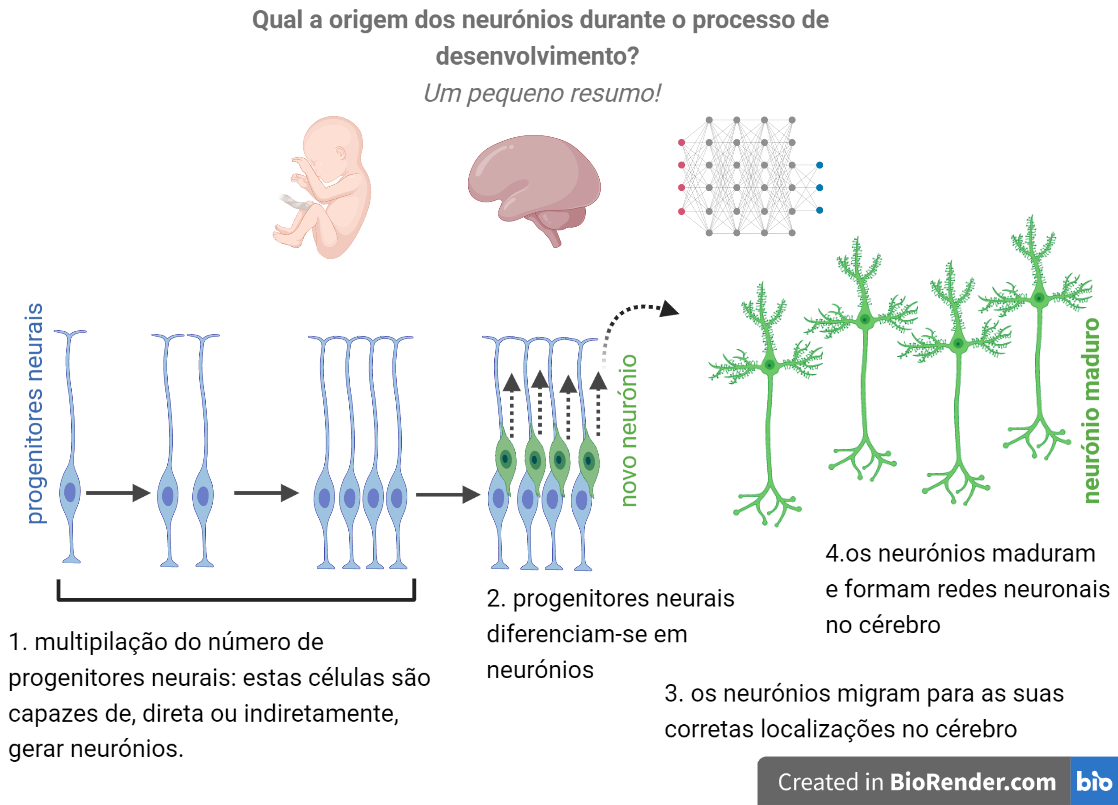
1. Ainda durante a gravidez, existem no cérebro embrionário um grupo de células denominadas progenitores neurais. Estas células darão origem à maioría das células do cérebro adulto. Inicialmente, estes progenitores neurais dividem-se muito rapidamente, multiplicando exponencialmente o seu número. Só mais tarde se diferenciam em neurónios, provavelmente as células mais conhecidas do cérebro.
2. Uma vez gerados, os neurónios migram, maduram e formam complexas redes no cérebro.
3. Uma vez a diferenciação ocorre, os neurónios já não são capazes de se dividir e, consequentemente, ter a capacidade de gerar novos neurónios. Isto significa que se, durante o desenvolvimento humano, os progenitores neurais sofrerem se diferenciarem em neurónios demasiado cedo, existirá uma perda precoce de progenitores neurais e, consequentemente, menos neurónios no cérebro adulto.
O que os investigadores descobriram é que, em organoides de cérebro gerados a partir de células de doentes com Síndrome de Rett, alguns progenitores neurais diferenciam-se em neurónios precocemente. Por outro lado, alguns tipos de neurónios demonstraram defeitos durante os processos de maturação e de migração até ao seu destino correto no cérebro.
Estes defeitos poderão contribuir para alguns dos sintomas apresentados durante o desenvolvimento por doentes com Síndrome de Rett.
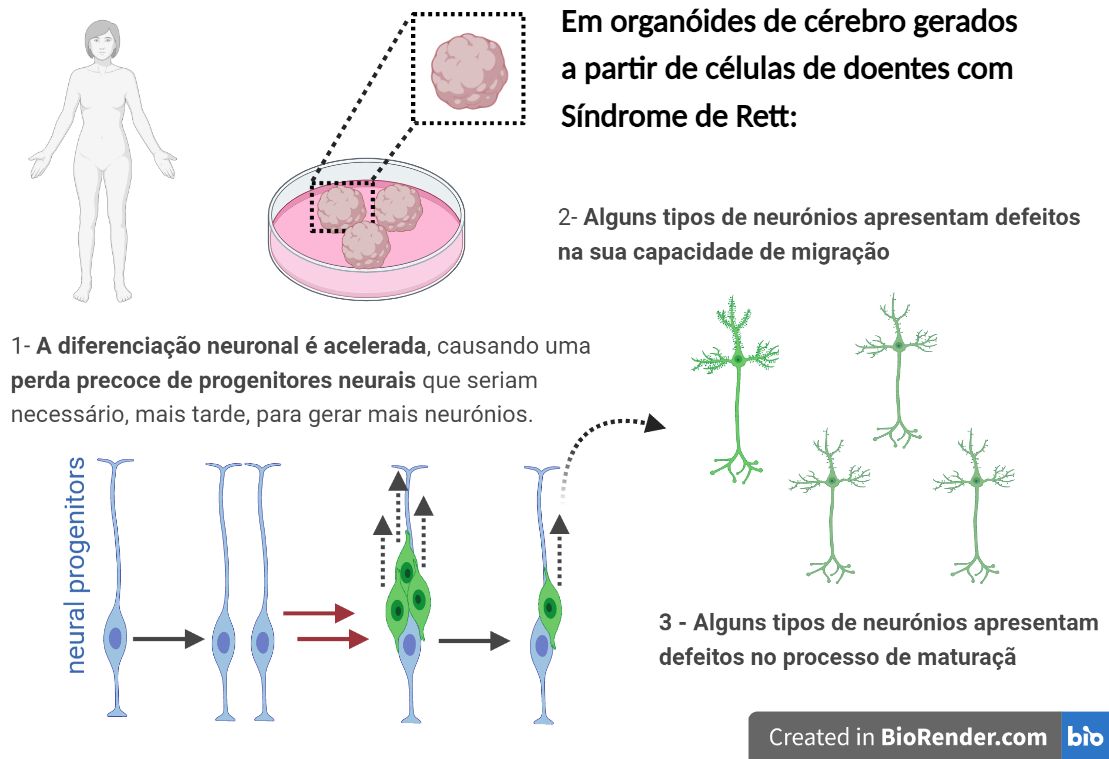
Para além de avanços mecanísticos na compreensão desta doença este estudo significa avanços técnicos no desenvolvimento de organoides de cérebro humano para o estudo de doenças raras como o Síndrome de Rett.
A acumulação deste tipo de conhecimento técnico é essencial para a utilização de organoides de cérebro humano nos processos de caracterização de doenças, medicina personalizada e teste de fármacos.
![]()
2. Artigo de investigação publicado na revista Neurobiology of Disease:
-Défice no sistema de adenosina no Síndrome de Rett: Novo alvo terapêutico para reforçar a sinalização de BDNF-
(Impairment of adenosinergic system in Rett syndrome: Novel therapeutic target to boost BDNF signalling)
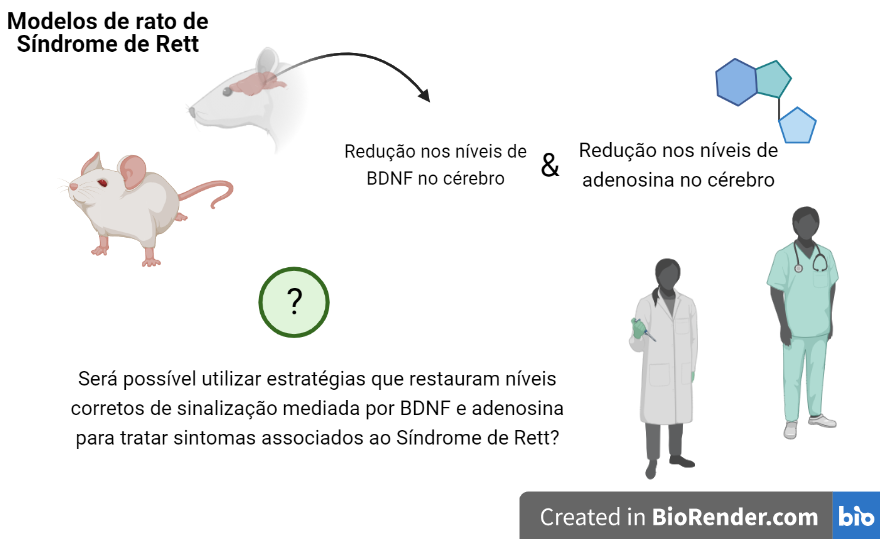
Outro destes estudos foi desenvolvido num projeto de colaboração entre investigadores do Hospital de San Joan de Deu, em Barcelona e o Instituto de Ciências Abel Salazar e o iMM, em Portugal.
Neste trabalho de investigação utilizaram-se modelos animais de Síndrome de Rett para estudar de que maneira os neurónios e a atividade neuronal se encontram afetados nesta doença.
Os investigadores estudaram qual o impacto em modelos de Síndrome de Rett de duas moléculas envolvidas no correto funcionamento dos neurónios. Uma dessas moléculas é o BDNF (factor de crescimento neuronal cujo nome tem origem no inglês, Brain Derived Nerve factor) e a adenosina.
O BDNF desempenha um papel crucial durante o desenvolvimento de neurónios. Está envolvido na capacidade destas células establecerem pontos de comunicação entre eleas no cérebro, as sinapses. Em linha com estudos anteriores, os investigadores verificaram que, neste modelo de Síndrome de Rett, existe uma redução no cérebro dos níveis de BDNF, bem como de outras proteínas envolvidas nesta via de sinalização celular.
Esta confirmação é muito importante: outros grupos de investigação mostraram anteriormente que, manipulando os níveis de BDNF no cérebro, é possível melhorar alguns dos sintomas em ratinhos modelo para Síndrome de Rett.
Translação direta deste conhecimento para o tratamento em humanos é ainda difícil.
No entanto, o que estes estudos demonstram é que moléculas que tenham a capacidade de potenciar os níveis ou a ação do BDNF no cérebro poderão representar um importante passo no desenvolvimento de novas estratégias terapêuticas.
Seguindo esta linha de raciocínio, os investigadores decidiram focar-se também na molécula adenosina.
ADENOSINA:
A adenosina é uma molécula que existe naturalmente no nosso cérebro.
A adenosina está envolvida na regulação da atividade dos neurónios em muitos processos biológicos como o controlo do sono. De facto, a adenosina funciona no cérebro como uma molécula antiepilética assegurando níveis corretos de atividade cerebral.
Vários estudos levados a cabo no iMM foram pioneiros ao demonstrar que, de facto, algumas das funções do BDNF dependem da adenosina.
O que os investigadores descobriram com este estudo é que, em modelos de ratinho para o Síndrome de Rett, existe uma redução dos níveis de adenosina no cérebro.
Esta é uma descoberta importante uma vez que existem grupos de investigação a testar a adenosina (e suas vias de sinalização) como um alvo terapêutico para o tratamento de crises epiléticas.
Consequentemente, um dos potenciais impactos deste trabalho poderá ser a possibilidade de utilizar estratégias baseadas na adenosina para o tratamento de doentes com Síndrome de Rett.

3. Artigo de investigação publicado na revista Neuroscience:
-Doenças raras do neurodesenvolvimento: manutenção do mistério ou utilização de uma ferramenta impressionante para investigação? – o caso do síndrome de Rett-
O cérebro é composto por cerca de 100 milhões de neurónios capazes de conduzir informação sob a forma de sinais elétricos. Estes neurónios organizam-se em redes complexas onde estabelecem conexões com outros neurónios. Estas conexões ocorrem em locais específicos designados de sinapses. Pensa-se que no cérebro adulto existirão mais de 125 triliões de sinapses!
-
- Nas sinapses, quando um neurónio transmite informação para outro neurónio, o sinal elétrico é convertido para um sinal químico através da libertação de neurotransmissores.
O reconhecimento destes sinais químicos pela outra célula ocorre, na maioria dos casos, na “parede” da célula (a membrana celular) e é mediado por proteínas conhecidas por recetores.
Nas sinapses, os neurónios têm também outras proteínas conhecidas por canais iónicos. Os canais iónicos controlam a passage de cargas eléctricas do interior para o exterior da célula e vice versa.
Quando um neurotransmissor se liga a um recetor específico, pode causar a abertura ou o fecho de canais iónicos, criando (ou bloqueando) a formação de um novo sinal eléctrico no segundo neurónio. 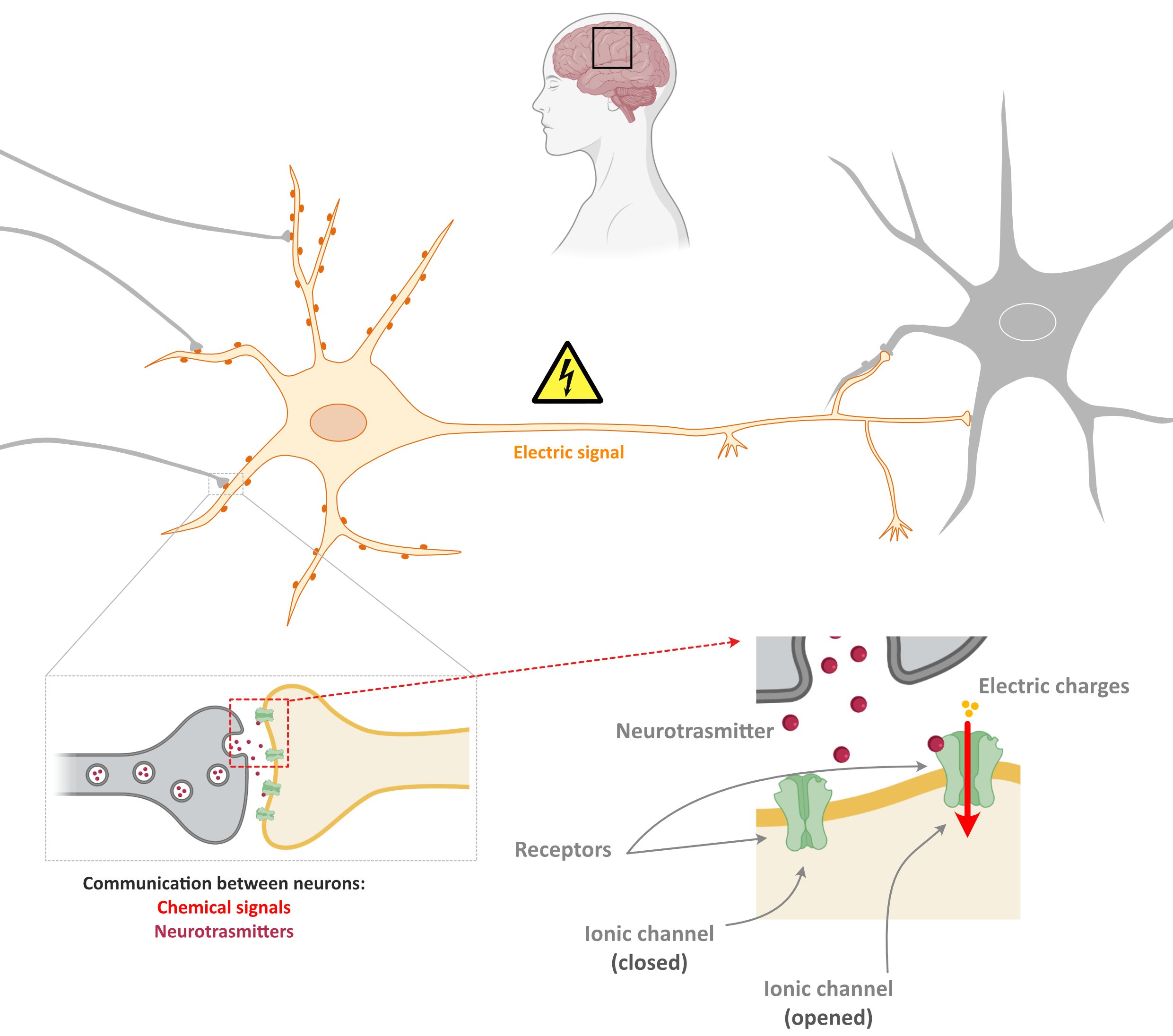
Este é o princípio através do qual a informação é transmitida no cérebro!
- Nas sinapses, quando um neurónio transmite informação para outro neurónio, o sinal elétrico é convertido para um sinal químico através da libertação de neurotransmissores.
Em várias doenças como o Síndrome de Rett, existe uma alteração no balanço entre os diferentes tipos de sinais, nomeadamente entre os sinais do tipo inibitório e excitatório, que pode levar à ocorrência de crises epiléticas.
Através de uma colaboração entre vários parceiros da rede EpiEpiNet, nomeadamente o iMM, a Amsterdam Medical Cenre e a Sapeinza University of Rome, foi utilizada uma técnica inovadora desenvolvida há vários anos por Ricardo Meledi no estudo do Síndrome de Rett. Neste estudo, os investigadores utilizaram a técnica desenvolvida por Ricardo Meledi para estudar o incorreto funcionamento de recetores inibitórios e de canais chave para o funcionamento do cérebro no contexto do Síndrome de Rett
...
Na EpiEpiNet acreditamos que, paso a paso, a comunidade científica alcançará um melhor conhecimento sobre as causas biológicas e sintomas do Síndrome de Rett e de outras doenças que envolvem o desenvolvimento de epilepsias refratárias.
Acreditamos que este conhecimento permitirá construir um caminho para o desenvolvimento de novas terapias, tão necessárias para o tratamento destas doenças.
Na EpiEpiNet temos orgulho de fazer parte deste processo.
Fevereiro 2021 - Mês das Doenças Raras

Existem mais de 300 000 pessoas em todo o mundo com doenças raras.
Por outro lado, foram já identificadas mais de 6000 doenças raras, cada uma com as suas características sintomáticas.
Algumas destas doenças raras estão associadas à ocorrência de crises epiléticas e, em alguns casos, ao aparecimento de epilepsias refratárias, resistentes ao tratamento com fármacos antiepiléticos.
Na nossa página partilhamos convosco informação sobre algumas destas doenças. Daremos uma atenção especial a novas descobertas que ajudam a compreender melhor os mecanismos celulares e moleculares por detrás de algumas destas doenças.
MAND
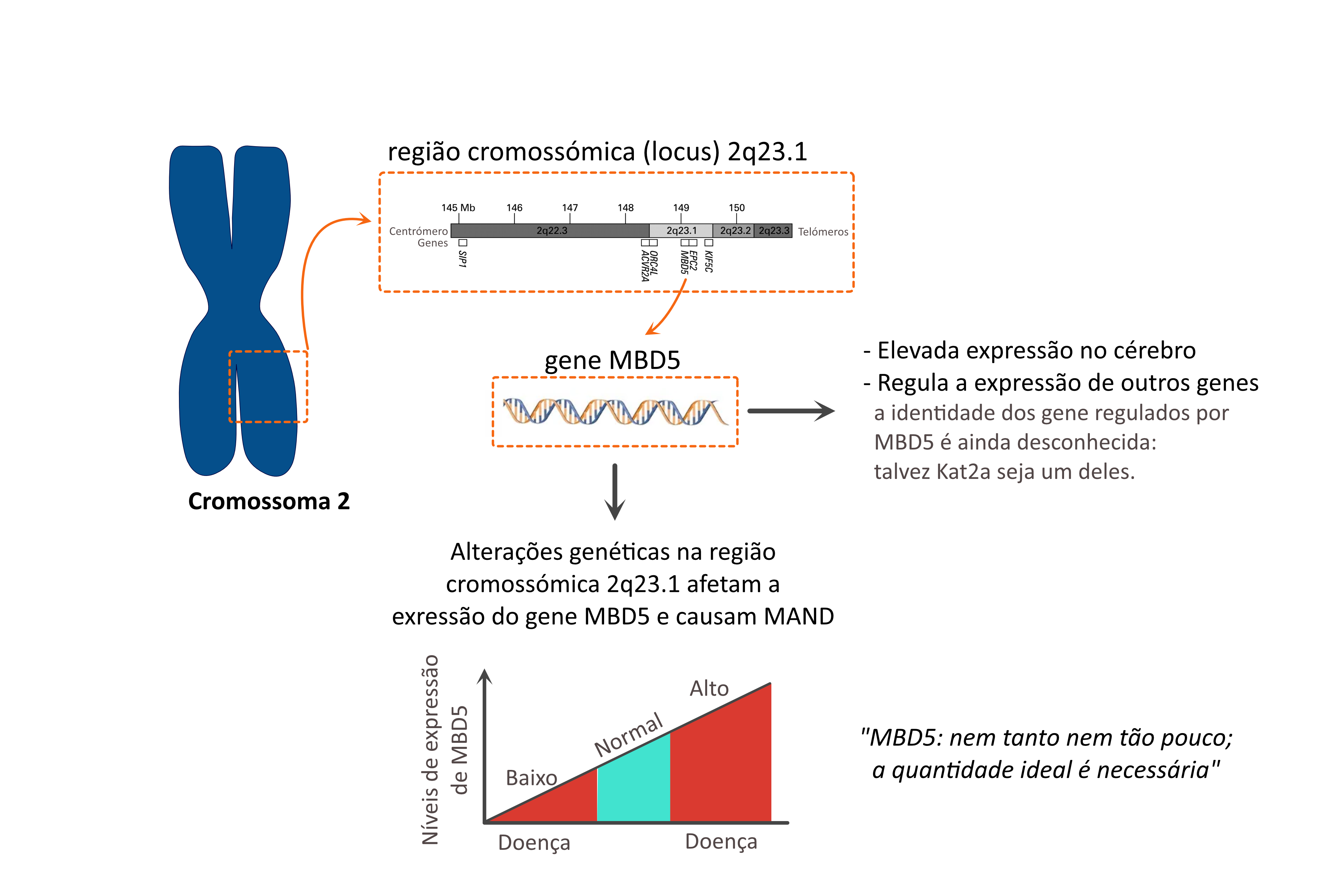
MAND é um termo utilizado para descrever um conjunto de doenças muito raras envolvendo alterações genéticas no uma região do cromossoma 2: o locus 2q23.1.
MAND tem um caráter dominante o que significa que basta que existam alterações genéticas numa das duas cópias do cromossoma 2 para que se desenvolva a doença.
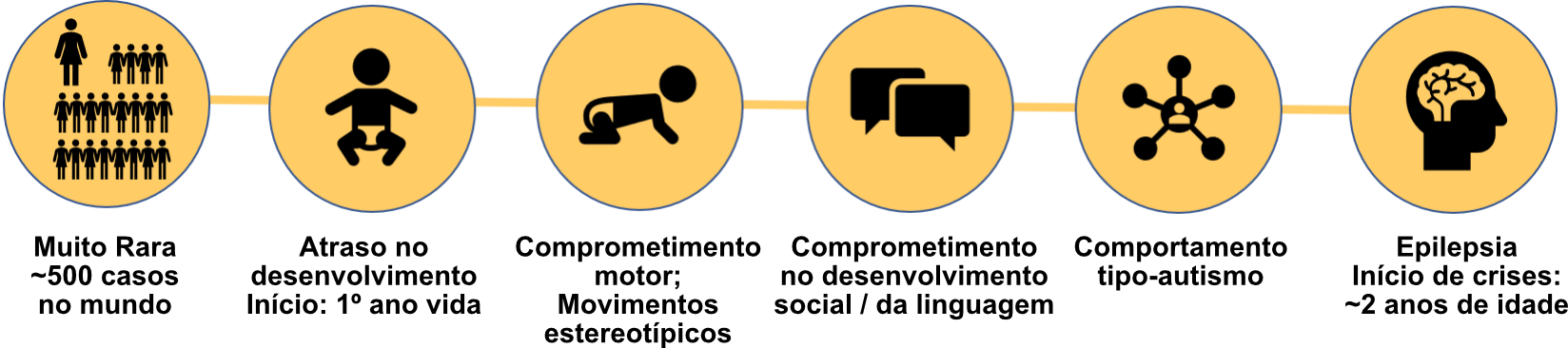
Em mais de 80% dos casos, MAND surge associado a:
- Um atraso no desenvolvimento com início durante o primeiro ano de vida.
Reflete-se em atrasos no desenvolvimento social e da linguagem. - Ocorrência de movimentos estereotípicos repetitivos
- Alterações no sono
- Microcefalia
- Padrões de comportamento muitas vezes semelhantes a autismo
- Crises epiléticas: o aparecimento das crises ocorre geralmente perto dos 2 anos de vida.
As crises epiléticas não foram bem caracterizadas nesta população. No entanto, sabe-se que a maioria dos indivíduos com MAND exibem só um tipo de crises epiléticas.
Alguns dos sintomas de MAND são semelhantes aos observados noutros Síndromes como Rett (ex. a repetição de movimentos estereotípicos com as mãos que se observa em doentes com Síndrome de Rett), Angelman, X-frágil, entre outros.
Lamentavelmente, o facto de que algumas das características de MAND se assemelham à de outros síndromes junto com o facto de se tratar de uma doença muito rara pode levar a um diagnóstico errado da doença.
Para lidar e tratar corretamente os doentes com MAND é muito importante adotar estratégias multidisciplinares e orientadas para cada indivíduo e que incluí áreas como: genética médica, terapia da fala e da linguagem, terapia física e ocupacional, desenvolvimento durante a infância, neurologia, nutrição e oftalmologia.
A informação sobre MAND e os mecanismos biológicos por trás da doença são escassos.
Ainda assim, nas últimas décadas a comunidade científica tem sido capaz de alcançar avanços na compreensão desta doença.
Aqui pode encontrar um bom resumo científico (escrito em inglês) feito por investigadores da Califórnia e do Texas, nos Estados Unidos sobre MAND:
https://www.nature.com/articles/ejhg201635
O termo MAND provem do inglês MBD5-associated neurodevelopmental disorder
(Distúrbio do neuro desenvolvimento associado a MBD5)
A origem de nome surge da observação de que alterações na região genética 2q23.1 causam alterações na expressão de MBD5: em alguns casos observa-se um aumento dos níveis da proteína MBD5; noutros casos, observa-se uma redução nos níveis de MBD5; quer o aumento quer a redução dos níveis de MBD5 está associada ao aparecimento de doença.
Este é um claro exemplo em biologia onde um controlo apertado dos níveis de expressão dos genes é essencial durante o desenvolvimento (nem demasiada expressão, nem pouca expressão; só o justo e necessário).
Mas porque é que a proteína MBD5 é tão importante?
- MBD5 é uma proteína que atua como regulador de transcrição.
Isto significa que tem como função regular a expressão de outros genes: algo semelhante à proteína MECP2 (envolvida no Síndrome de Rett). De facto, MBD5 e MECP” pertencem à mesma família de proteínas.
No entanto, quais os genes que são regulados pela MBD5 é ainda uma incógnita.- Furthermore, high levels of MBD5 are found in the brain.
- Por outro lado, sabe-se que os níveis de MBD5 no cérebro são particularmente elevados.
Para compreender os mecanismos biológicos por trás de doenças como MAND e desenvolver terapias apropriadas é importante contar com modelos animais que permitam aos investigadores estudar estas doenças.
De facto, nos últimos anos vários modelos animais para MAND têm sido criados.
Estes modelos animais utilizam estratégias de engenharia genética para alterar os níveis de MBD5 em ratos. Sabe-se agora que, em alguns casos, estes modelos mimetizam alguns dos sintomas e características observados durante o desenvolvimento em doentes com MAND.
Apesar de ser necessária uma melhor caracterização destes modelos, só por si, a existência sua existência já significa boas notícias uma vez que são uma potente ferramenta para a compreensão de MAND.
Outra questão que necessita ser clarificada pelos investigadores é se MBD5 é a única proteína afetada em doentes com MAND.
Por exemplo, na proximidade da região 2q23.1 (envolvida em MAND) encontram-se genes que codificam para outras proteínas como ORC4, KIF5C e EPC2. Sabe-se que estas proteínas desempenham também um papel importante durante o desenvolvimento e funções cerebrais.
No entanto, é ainda uma incógnita se estas proteínas também contribuem para algumas das características de MAND.
…
Na EpiEpiNet acreditamos que, paso a paso, a comunidade científica alcançará um melhor conhecimento sobre as causas biológicas e sintomas de doenças raras que envolvem o desenvolvimento de epilepsias refratárias.
Acreditamos que este conhecimento permitirá construir um caminho para o desenvolvimento de novas terapias, tão necessárias para o tratamento destas doenças.
Na EpiEpiNet temos orgulho de fazer parte deste processo.